

Escrever bem não é apenas uma habilidade técnica — é um ato de comunicação...
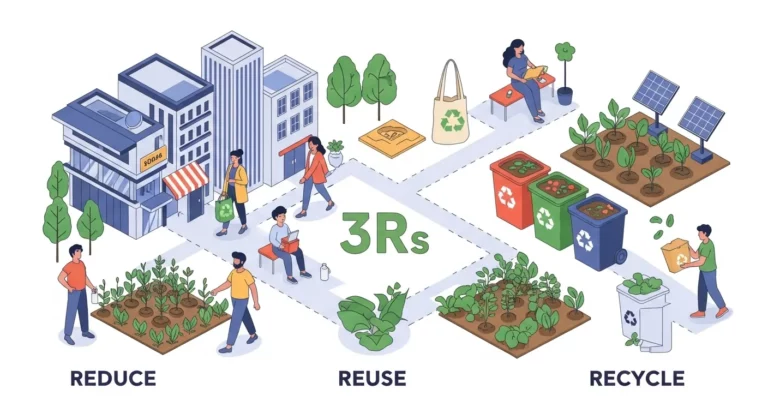
Quando pensamos em sustentabilidade, uma das primeiras imagens que surge é a da coleta...

O folclore brasileiro é um mosaico vivo, pulsante e diverso, resultado da interação histórica...
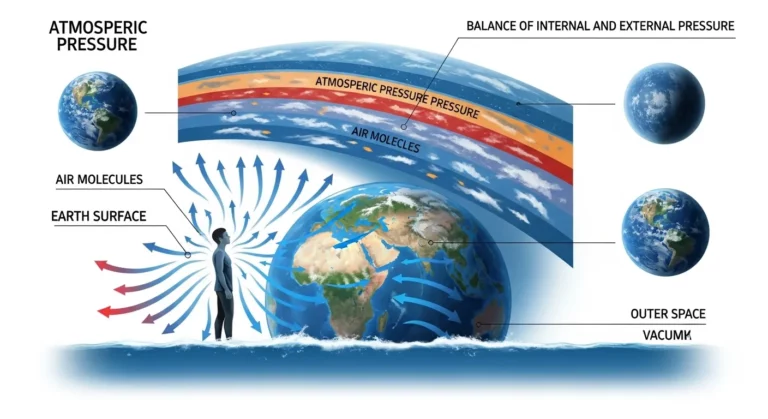
Você já parou para pensar que está sendo pressionado por toneladas de ar neste...

A saúde não é construída apenas em hospitais, clínicas ou laboratórios. Ela nasce, principalmente,...

O avanço da tecnologia digital transformou profundamente a maneira como aprendemos, ensinamos e interagimos...

Vivemos em uma era marcada pelo consumo. A cada dia, somos incentivados a comprar...

Falar de energias renováveis é falar de vida, de futuro e de responsabilidade coletiva....

O ensino do folclore ocupa um espaço privilegiado na educação, especialmente em países culturalmente...

A sociedade atual vive cercada por informações. Notícias chegam pelo celular, pela televisão, pelas...